Import your data
Expression file
Upload in DIANE the raw counts that were obtained after the bioinformatic pipeline of mapping and quantification of your reads. It gives, for each gene, the transcript abundance found in each of your experimental condition and replicates.
The needed matrix-like file shoud contain a column named “Gene”, containing all your gene IDs.
The other columns should be your sample names, noted as follow : conditionName_replicate (see example below). For example, HM_2 corresponds to the second replicate of the experimental condition HM,
In order for your input to be compatible with the proposed organisms, it should contain gene IDs as follow :
- For Arabidopsis thaliana : TAIR IDs (ex: AT1G01020, or AT1G01020.1)
- For Human : ensembl IDs (ex: ENSG00000005513)
- For Mus musculus : ENSMUSG00000087910
- For Drosophilia : FBgn0000036
- For C. elegans : WBGene00000042
- For Lupinus albus : Lalb_Chr00c02g0404151
- For E-coli : acpS
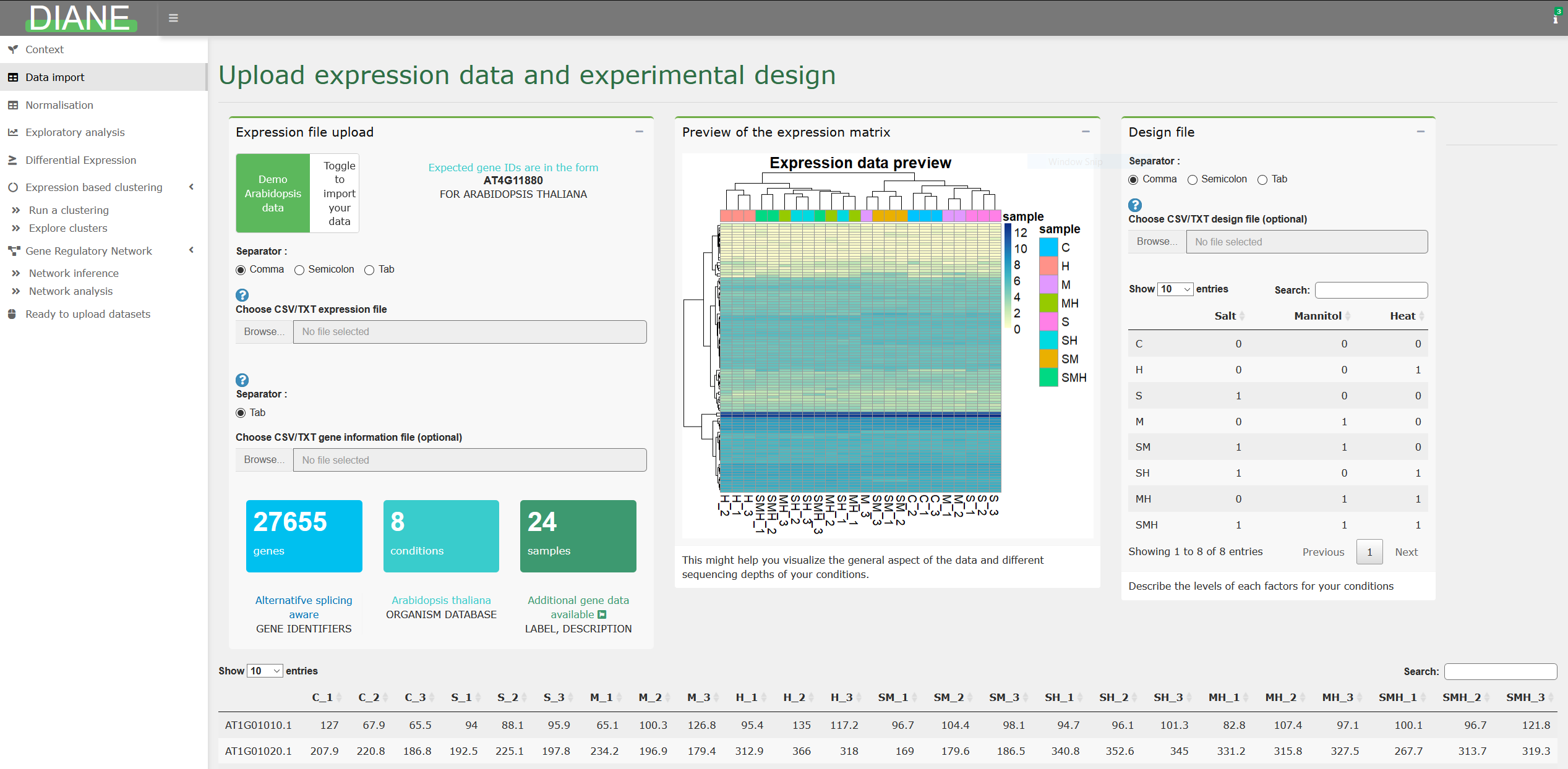
Here is the head of our companion expression file, containing 24 Arabidopsis thaliana’s samples :
Gene,C_1,C_2,C_3,S_1,S_2,S_3,M_1,M_2,M_3,H_1,H_2,H_3,SM_1,SM_2,SM_3,SH_1,SH_2,SH_3,MH_1,MH_2,MH_3,SMH_1,SMH_2,SMH_3
AT1G01010.1,127.0,67.9,65.5,94.0,88.1,95.9,65.1,100.3,126.8,95.4,135.0,117.2,96.7,104.4,98.1,94.7,96.1,101.3,82.8,107.4,97.1,100.1,96.7,121.8
AT1G01020.1,207.9,220.8,186.8,192.5,225.1,197.8,234.2,196.9,179.4,312.9,366.0,318.0,169.0,179.6,186.5,340.8,352.6,345.0,331.2,315.8,327.5,267.7,313.7,319.3
AT1G01030.1,32.7,34.4,55.8,33.6,15.9,31.3,21.4,29.8,33.5,47.7,39.0,51.1,25.2,31.2,34.4,61.0,65.5,61.2,45.0,36.2,55.9,46.0,42.7,56.8Expression values should be zeros or positive integers.
Ideally, the expression file should not have been normalized in any way. However, it is possible in DIANE to skip normalization if you have no choice but to upload normalized counts.
Design
If desired, you can specify the experimental design of your data. This will be used only for Generalized Poisson regression in cluster characterization, so this is optional.
To do so, you must provide, for each condition name, the level of the factors corresponding to that condition in your study.
Let’s take our companion data as an example. Here, the experiment we chose for our demo includes 3 factors, heat stress, mannitol stress, as well as salinity stress.
You should first identify which level of each factor can be considered as the control level, and which is a perturbation. In our demo, the control condition is referred to as C. Then, the perturbation are specified by their corresponding letter, from simple to triple stress combination. For example, SH corresponds to salt and heat stresses in the control level of mannitol. As a consequence, its levels would be 1,0,1.
The design file is thus a matrix with condition names as rows, and factor names as columns. It should contain a column named “Condition”, as you can see in our example below :
Condition,Salt_stress,Mannitol_stress,Heat_stress
C,0,0,0
H,0,0,1
S,1,0,0
M,0,1,0
SM,1,1,0
SH,1,0,1
MH,0,1,1
SMH,1,1,1Many RNASeq anlysis only study one factor, which would not be a problem to use DIANE. If you have two conditions named control and trt, the design would contain only one factor column:
Condition,treatment
contr,0
trt,1Organisms
A number of model organisms are already stored in the application, so that, if your expression file contains the expected gene IDs, an annotation will automatically be found and displayed along the pipeline. This includes gene labels, description, ontologies and transcriptional regulators.
Else, you can upload those information for your organism.
You can find a number of pre-formatted datasets in the last tab of the online application at https://diane.bpmp.inrae.fr, that meet the expected format and type of gene IDs.